本文研究了質子交換膜燃料電池(PEMFC)中碳材料的潤濕性。PEMFC是一種將氫氣和氧氣的化學能轉換為電能的裝置,其性能受催化劑層(CL)中水管理的影響很大。碳材料作為PEMFC中催化劑鉑納米粒子的載體,其潤濕性對CL中的水分布和傳輸至關重要。本文合成了膠體印跡碳(CIC)和有序介孔碳(OMC)等碳材料,并通過熱處理和表面氟化等方法調控其潤濕性,采用接觸角動力學(CAK)和水蒸氣吸附(WVS)等方法評估了這些材料的潤濕性。研究發現,熱處理和表面氟化均能有效提高碳材料的疏水性,而CIC由于其較大的孔徑和堅固的孔壁,在PEMFC應用中顯示出更大的潛力。
隨著全球能源需求的增長,尋找可持續的能源解決示意圖變得尤為重要。PEMFC作為一種高效、清潔的能源轉換技術,近年來受到了廣泛關注。然而,PEMFC的商業化仍面臨成本高和耐久性差等挑戰。
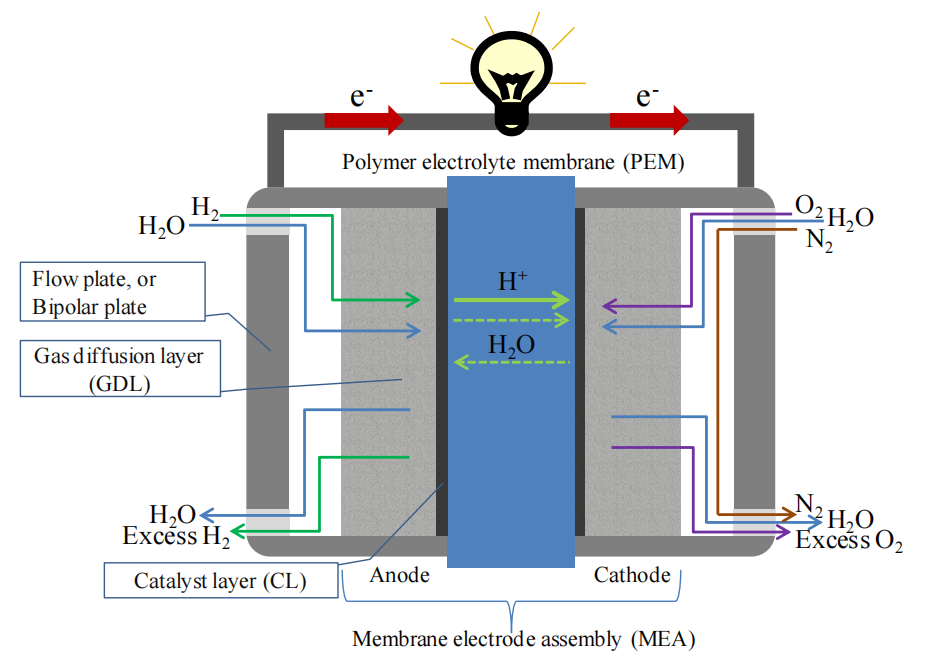
圖1.1. 氫-空氣型質子交換膜燃料電池(PEMFC)截面結構示意圖。
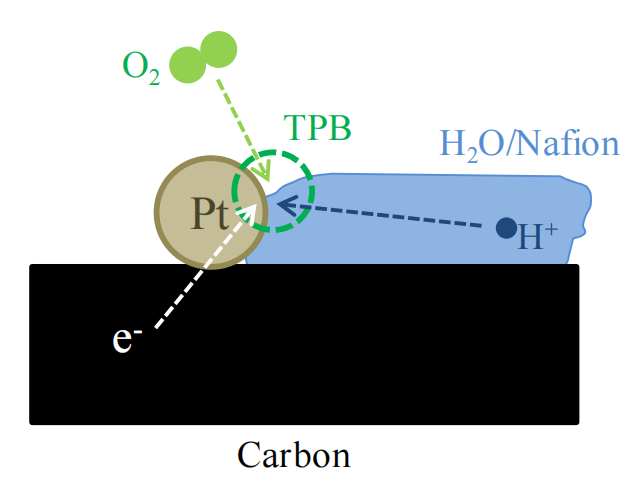
圖1.2. 質子交換膜燃料電池(PEMFC)陰極中發生氧還原反應(ORR,反應1.2)的三相邊界(TPB)示意圖。
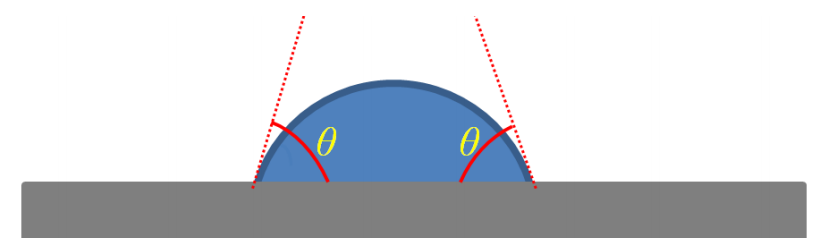
圖1.3. 水滴(藍色)在平坦樣品表面(灰色)的接觸角(θ)示意圖。
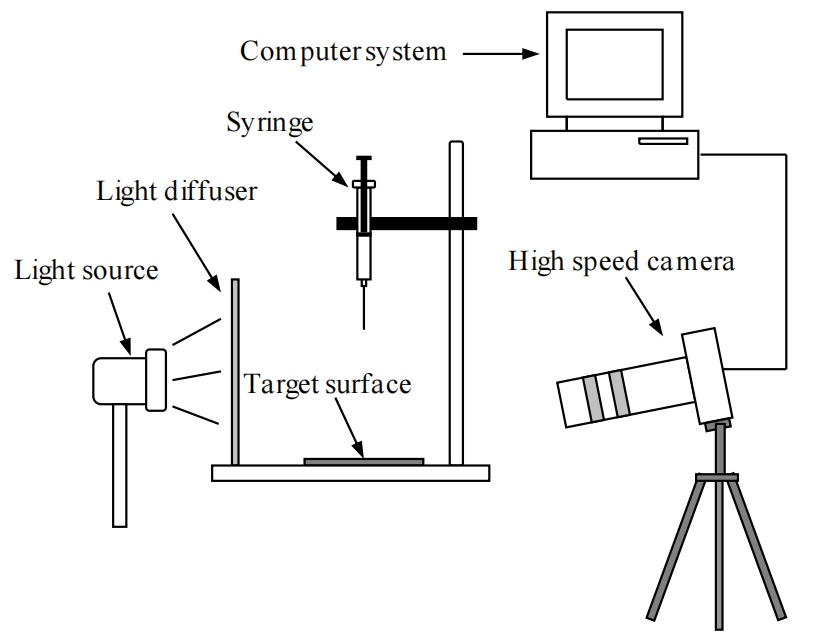
圖2.1. 用于液滴沖擊實驗的實驗裝置示意圖。
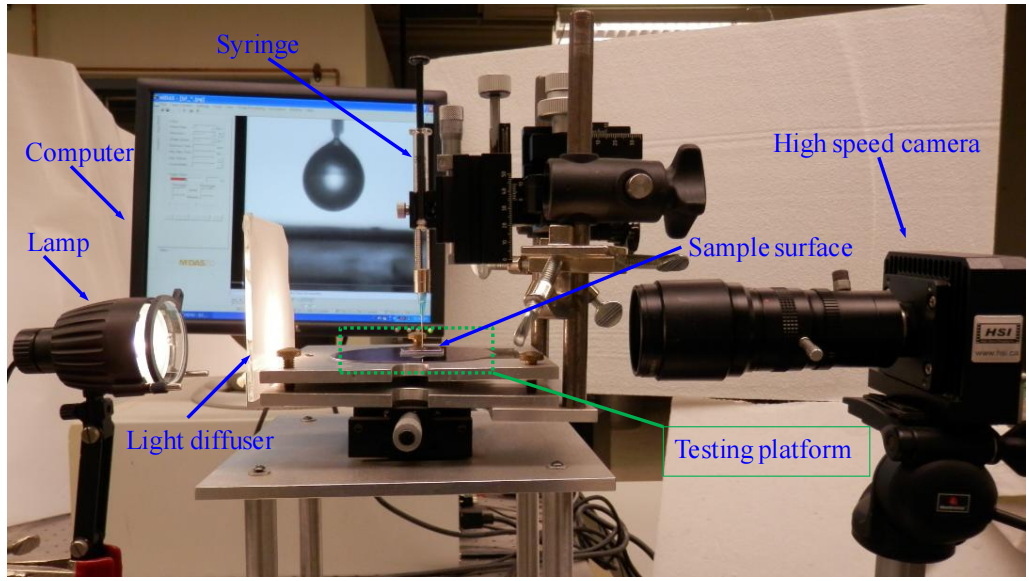
圖2.2. 用于接觸角動力學(CAK)測量的實驗裝置照片。

圖2.3. (a) 用于水蒸氣吸附(Water Vapor Sorption,簡稱WVS)測量的密封干燥器照片,其中碳樣品置于坩堝內;(b) 干燥器內坩堝的俯視照片(空坩堝作為對照實驗使用)。在將樣品放入干燥器前一天,已向密封干燥器底部加入100毫升蒸餾水。
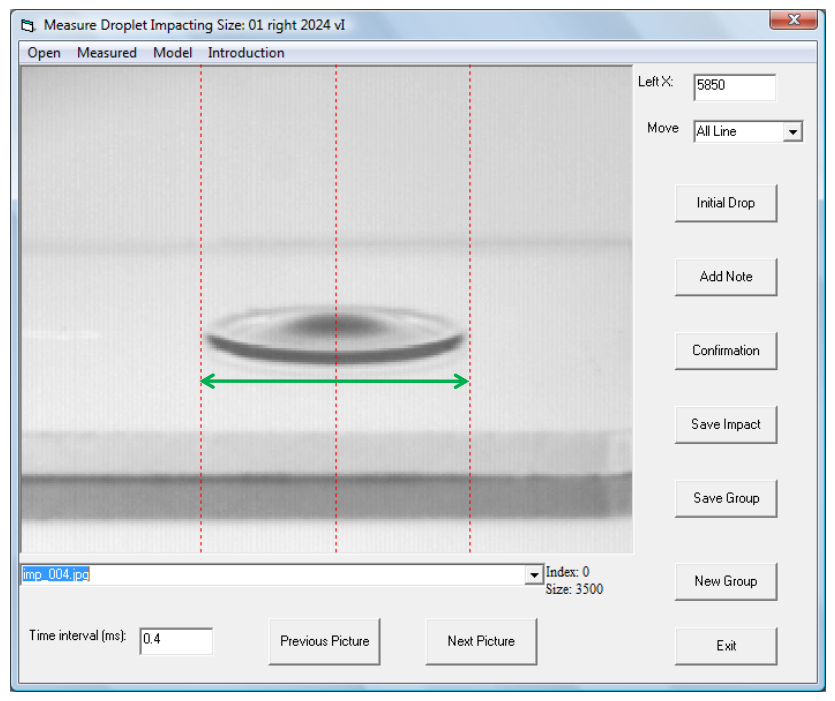
圖2.4. 用于測量液滴在樣品表面瞬時鋪展直徑(綠色箭頭)的Visual Basic軟件界面。
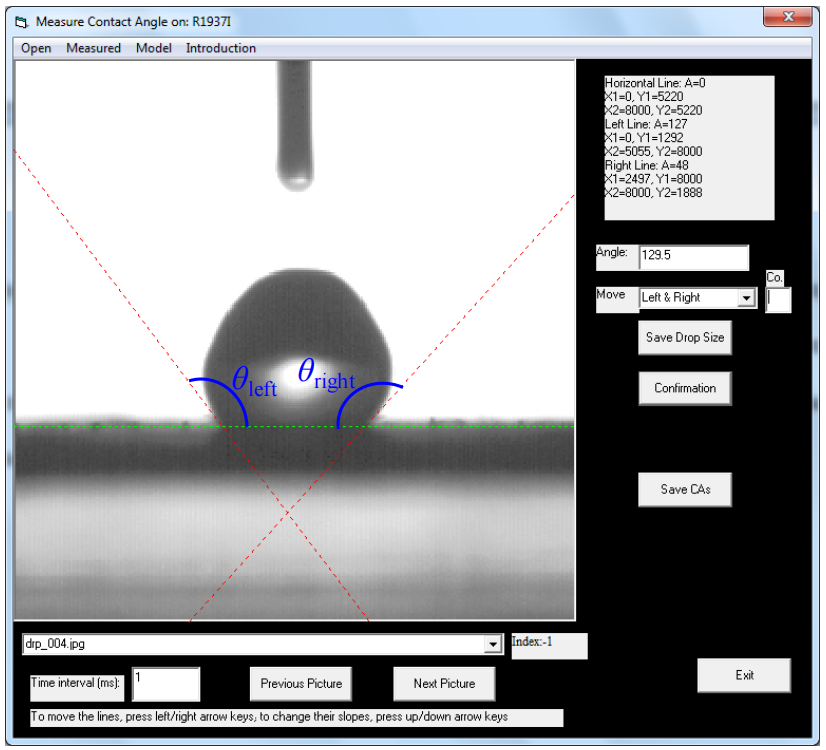
圖2.5. 用于測量水在壓實碳片上接觸角的Visual Basic軟件界面。
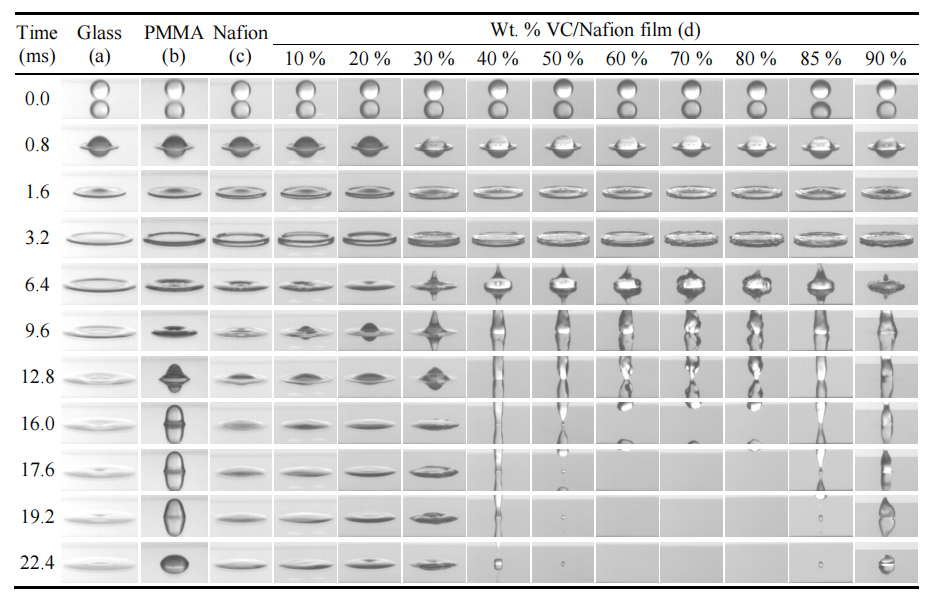
圖3.1. 直徑2.4 mm水滴撞擊不同基底材料的動態序列圖像:(a) 裸玻璃;(b) 聚甲基丙烯酸甲酯(PMMA);(c) 全氟磺酸樹脂(Nafion);(d) 玻璃基體表面涂覆的10–90 wt.% 火山碳(VC)/Nafion復合薄膜(沖擊高度均為120 mm)。液滴行為的差異源于材料表面能特性差異。
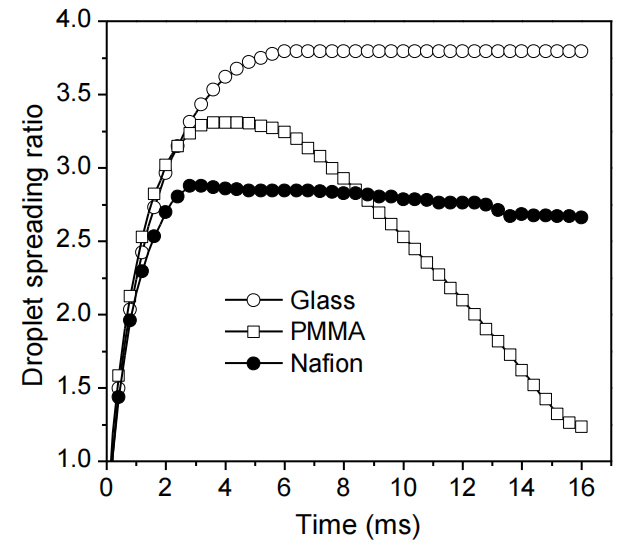
圖3.2. 水滴撞擊裸玻璃、聚甲基丙烯酸甲酯(PMMA)及全氟磺酸樹脂(Nafion)表面的實驗鋪展率(公式3.1)
 圖3.3. Nafion薄膜表面與截面的掃描電鏡(SEM)表征及結構示意圖
圖3.3. Nafion薄膜表面與截面的掃描電鏡(SEM)表征及結構示意圖
(a) 玻璃基底上旋涂1%全氟磺酸樹脂(Nafion)/異丙醇溶液制備的薄膜俯視表面Pd/Au濺鍍層SEM圖像。圖中灰色顆粒(可能為玻璃碎屑)用于輔助聚焦13;(b) 薄膜截面SEM圖像;(c) 本工作中Nafion薄膜表面結構示意圖,深色球體代表納米通道開口;(d) 薄膜截面結構示意圖,垂直深色條帶對應納米通道分布。
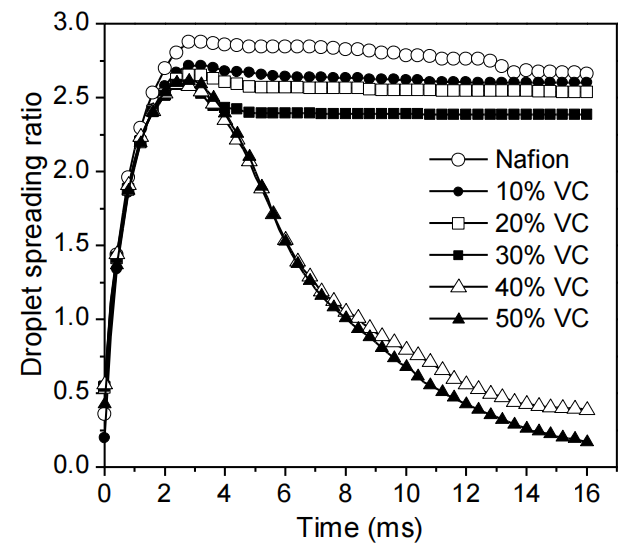
圖3.4. 水滴撞擊旋涂全氟磺酸樹脂(Nafion)薄膜及系列火山碳(VC)/Nafion復合基底表面(10–50 質量分數)的實驗鋪展率。
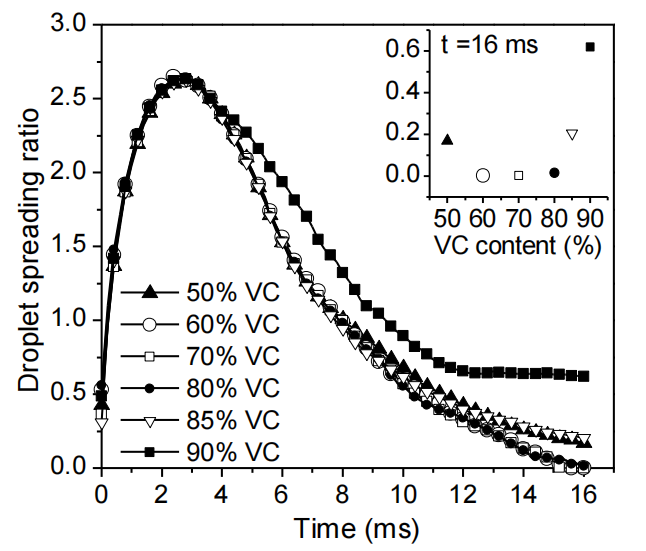
圖3.5. 水滴撞擊火山碳(VC)/全氟磺酸樹脂(Nafion)復合基底表面(50–90 質量分數)的實驗鋪展率。插圖為 t = 16 毫秒時刻的鋪展率數據。
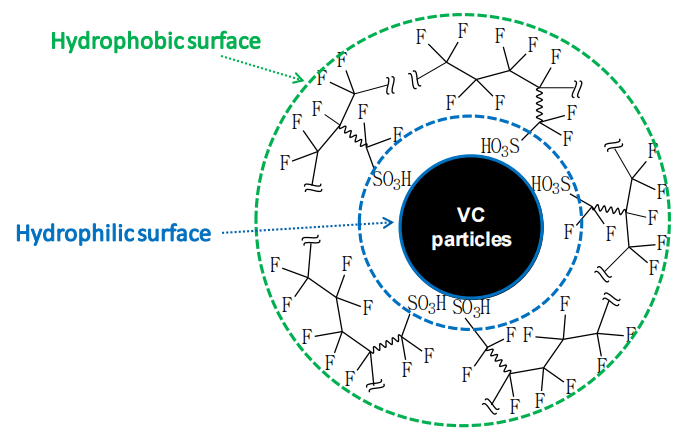 圖3.6. 全氟磺酸樹脂(Nafion)在火山碳(VC)顆粒表面的取向結構示意圖
圖3.6. 全氟磺酸樹脂(Nafion)在火山碳(VC)顆粒表面的取向結構示意圖
圖中顯示Nafion疏水性主鏈朝外排列,從而形成疏水性外表面。

3.7. 圖3.7鈀/金濺射處理的火山碳(VC)/全氟磺酸樹脂(Nafion)復合基底表面掃描電子顯微鏡(SEM)圖像。(a–b) 20% VC/Nafion、(c–d) 40% VC/Nafion、(e–f) 60% VC/Nafion、(g–h) 80% VC/Nafion、(i–j) 90% VC/Nafion。第一列與第二列分別為低倍率與高倍率下的表面形貌。
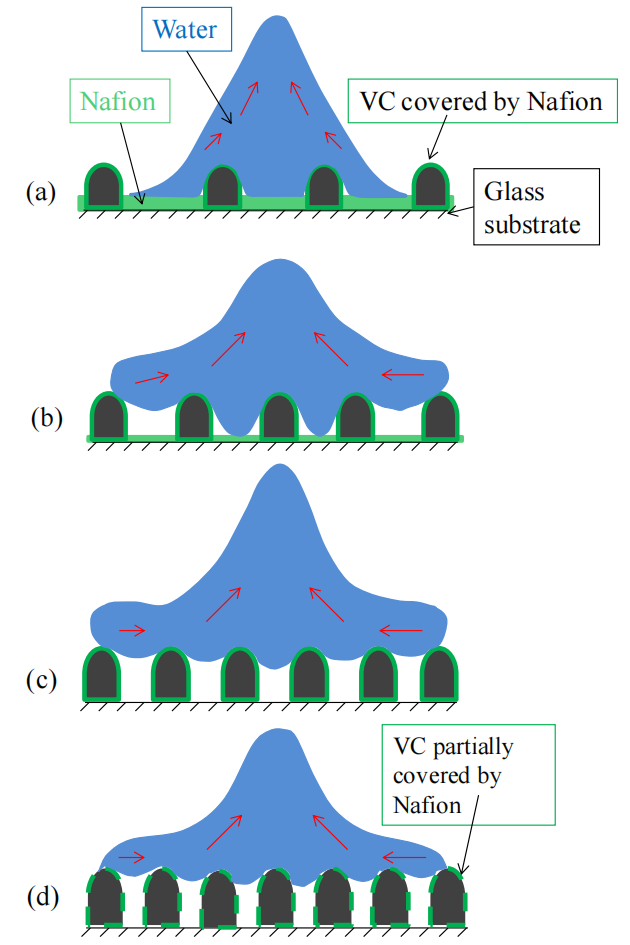
圖3.8. 水滴(藍色)撞擊不同比例火山碳(VC)/全氟磺酸樹脂(Nafion)復合材料表面后的回縮過程示意圖。(a) 30% VC/Nafion、(b) 40% VC/Nafion、(c) 60% VC/Nafion、(d) 90% VC/Nafion。圖中捕捉了水滴在 t = 6.4 毫秒時的動態(如圖3.1所示),展示了水滴與碳基/Nafion復合表面間的相互作用。紅色箭頭表示水滴的運動方向。
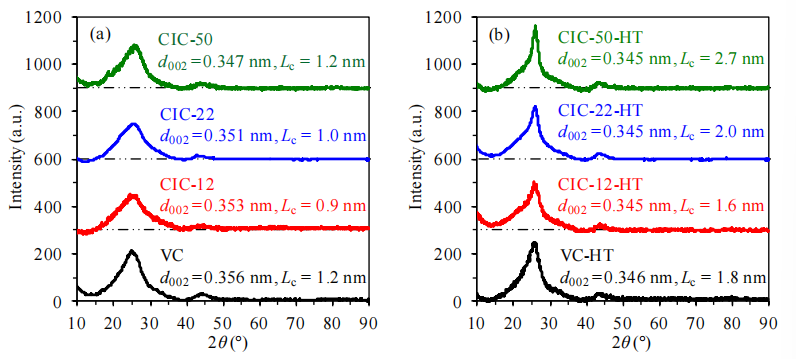
圖4.1. 碳材料樣品熱處理前后的X射線衍射(XRD)圖譜。(a) 未熱處理樣品、(b) 氮氣氣氛中1500°C熱處理(HT)后樣品。曲線下方的點劃虛線表示各圖譜的基線。通過對應圖譜中2θ≈25°處的衍射峰,應用布拉格定律計算層間距(d<sub>002</sub>),并基于謝樂方程獲得c軸方向晶粒尺寸(L<sub>c</sub>)[164]。

圖4.2. 火山碳(VC)及其熱處理衍生物與不同比例碳離子液體復合材料的場發射掃描電子顯微鏡(FE-SEM)高倍圖像。(a) VC、(b) VC-HT(熱處理)、(c) CIC-12、(d) CIC-12-HT、(e) CIC-22、(f) CIC-22-HT、(g) CIC-50、(h) CIC-50-HT。所有粉末樣品均以碳導電膠帶為基底,于高倍率下觀測。

圖4.3. 不同碳材料粉末的低倍率場發射掃描電子顯微鏡(FE-SEM)圖像。(a) 火山碳(VC)、(b) VC經熱處理(VC-HT)、(c) CIC-12、(d) CIC-12-HT、(e) CIC-22、(f) CIC-22-HT、(g) CIC-50、(h) CIC-50-HT。所有樣品均以碳導電膠帶為基底,于低倍率下觀測。
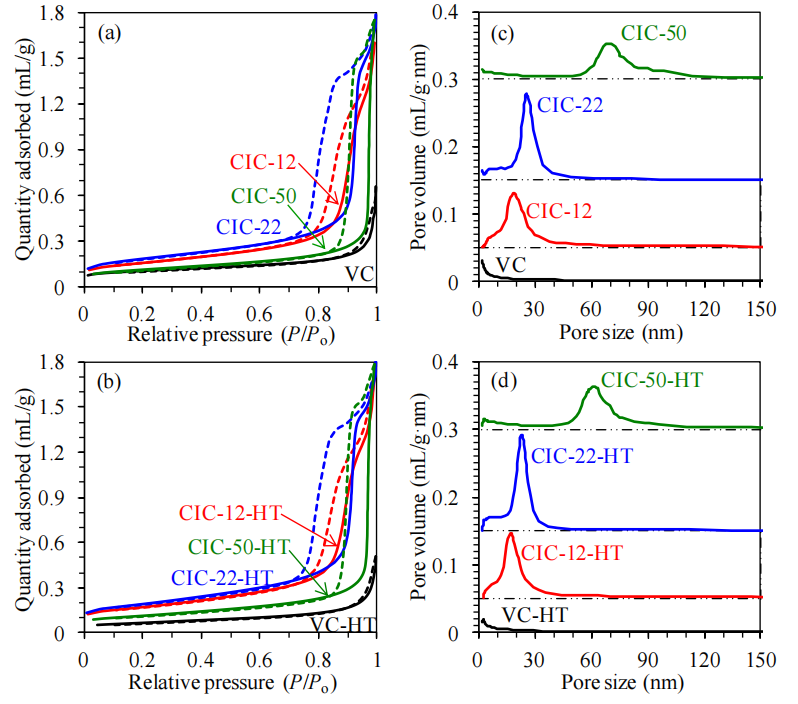
圖4.4. 熱處理前后碳材料的氮氣吸脫附曲線與孔徑分布分析。(a) 未熱處理、(b) 氮氣氣氛中1500°C熱處理2小時(HT)后的碳材料氮氣吸附(實線)與脫附(虛線)曲線。基于吸附支數據,采用Barrett-Joyner-Halenda(BJH)方法計算孔徑分布(c和d),并以碳黑t曲線為標準確定吸附氮膜的統計厚度[172]。分布圖(c和d)中的點劃虛線表示基線。
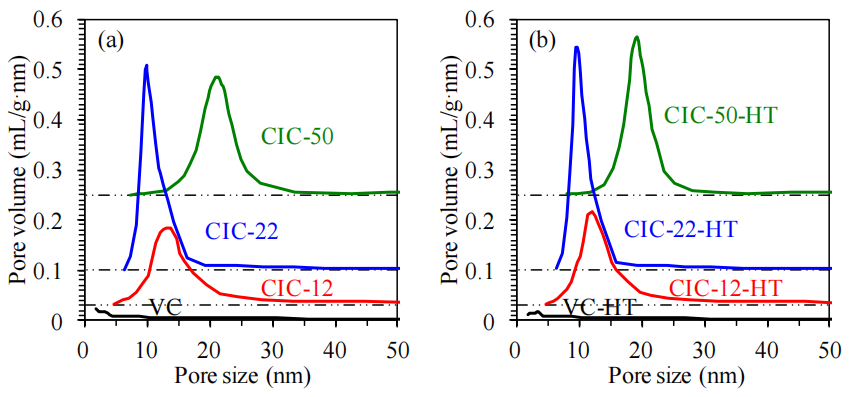
圖4.5. 熱處理前后碳材料的孔徑分布對比。(a) 未熱處理、(b) 在氮氣氣氛中于1500°C下熱處理2小時(HT)后的碳材料孔徑分布。基于氮氣吸脫附等溫線(圖3)的脫附支數據,采用Barrett-Joyner-Halenda(BJH)方法計算,并以碳黑t曲線為標準確定吸附氮膜的統計厚度[172]。分布曲線下方的點劃虛線(dash-dot-dot lines)表示對應圖的基線。
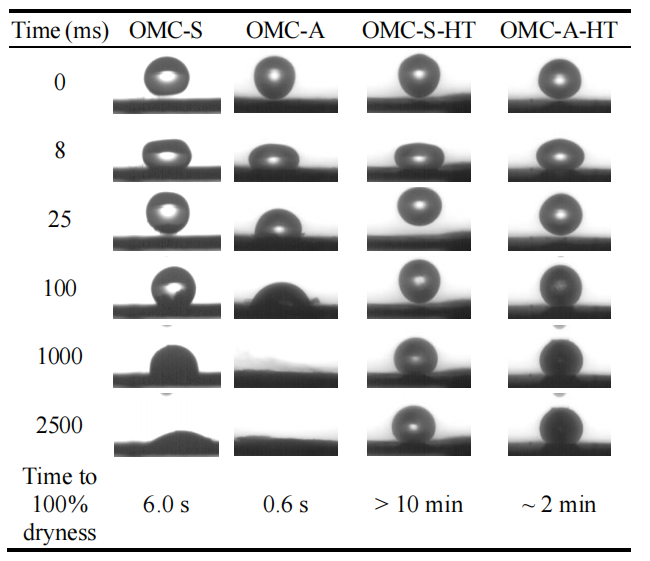
圖4.6 展示了水滴(直徑約2.7毫米)在平坦碳素顆粒壓片表面的一系列連續圖像,記錄了該樣品在氮氣氣氛下經1500°C熱處理前后的變化過程,顯示了水在壓片表面接觸角動力學(CAK)特征。文中所指"估計的完全干燥時間"表示碳素顆粒壓片完全吸收沉積在其表面的水滴所需時長。
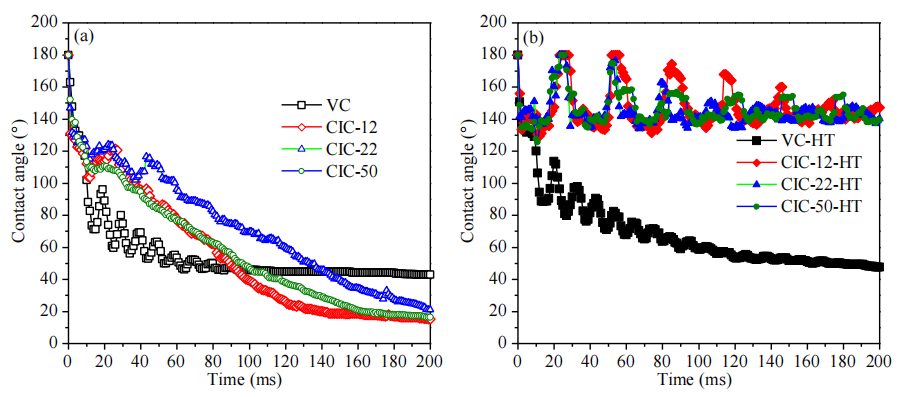
圖4.7. 碳顆粒表面水滴接觸角動力學(CAK)分析。(a) 未熱處理、(b) 在氮氣氣氛中于1500°C下熱處理后的碳顆粒表面水滴沉積后的接觸角動態變化。
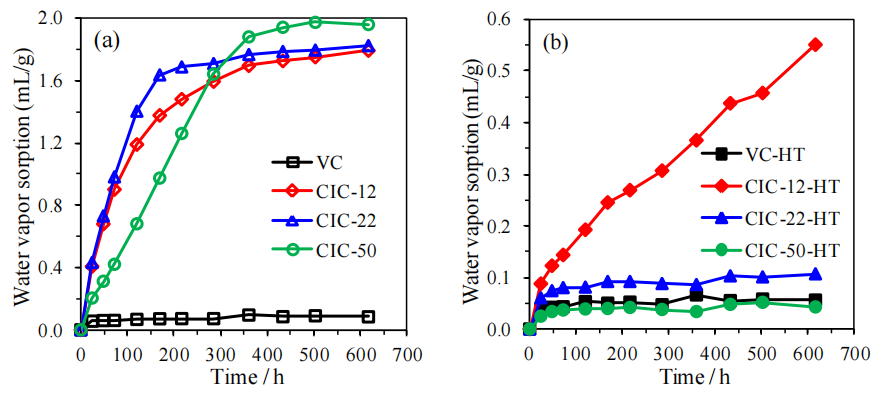
圖4.8. 熱處理前后碳材料的水蒸氣吸附(VWVS)對比。(a) 未熱處理、(b) 在氮氣氣氛中于1500°C下熱處理后的碳材料在室溫下的水蒸氣吸附性能。
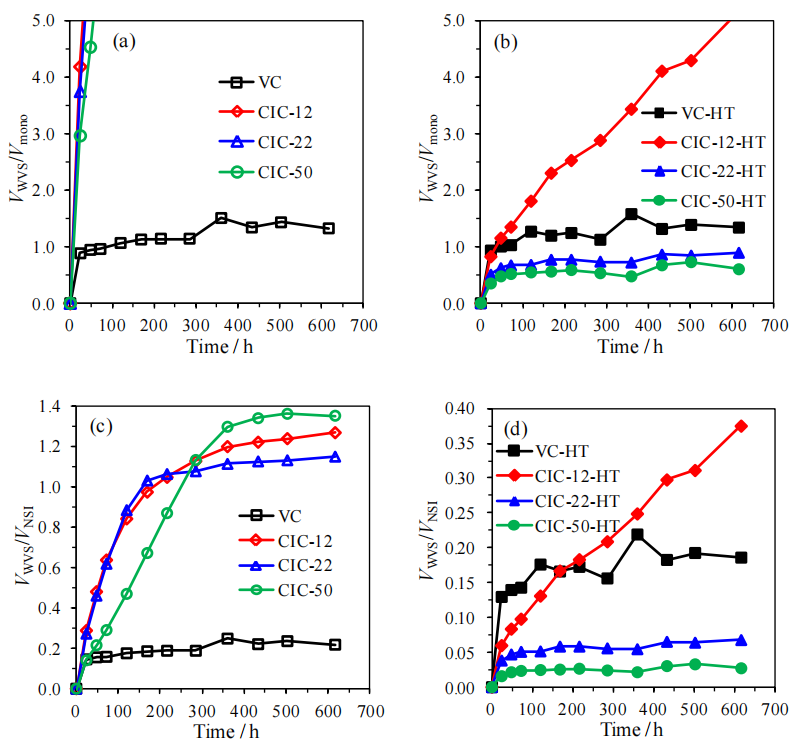
圖4.9. 碳材料歸一化水蒸氣吸附數據(基于圖4.8)(a) 未熱處理、(b) 在氮氣氣氛中于1500°C下熱處理后的碳材料水蒸氣吸附數據。基于氮氣吸附等溫線(圖4.4)所得表面積(表4.2)與孔體積(表4.3)進行歸一化處理。
(注:數據歸一化依據:以氮氣吸附法測得的比表面積和孔體積為基準,消除材料物理結構差異對吸附性能的影響;
測試條件標注:
熱處理溫度(°C)與氣氛(氮氣)標注符合實驗參數規范;
方法一致性:
歸一化處理邏輯與圖4.8中吸附數據來源保持一致。)
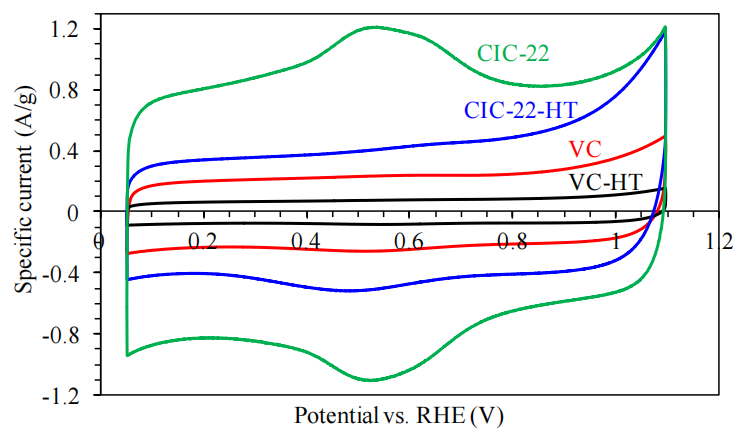
圖4.10. 不同碳材料的循環伏安(CV)性能對比。VC(未熱處理)、VC-HT(熱處理)、CIC-22(未熱處理)及CIC-22-HT(熱處理)在氮氣飽和的0.5 M H<sub>2</sub>SO<sub>4</sub>水溶液中的循環伏安曲線,掃描速率為10 mV/s。
(注:基于電化學測試規范。
測試條件標注:
電解液為氮氣飽和0.5 M硫酸溶液,避免氧還原反應干擾;
掃描速率(10 mV/s)標注符合電化學參數規范;
樣品命名邏輯:
后綴“HT”表示經1500°C氮氣氣氛熱處理后的樣品,與圖4.7-4.9命名保持一致;
電化學性能關聯性:
熱處理可能通過優化碳材料導電性、孔隙結構或表面官能團分布,影響其電容特性與氧化還原響應;
圖表可比性:
四組樣品在相同電解液與掃描速率下測試,確保電化學行為差異的可比性。)
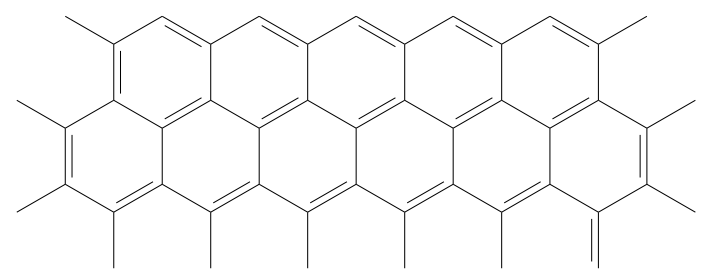
示意圖4.1. 石墨烯層頂面鋸齒形邊緣構型模型[191]
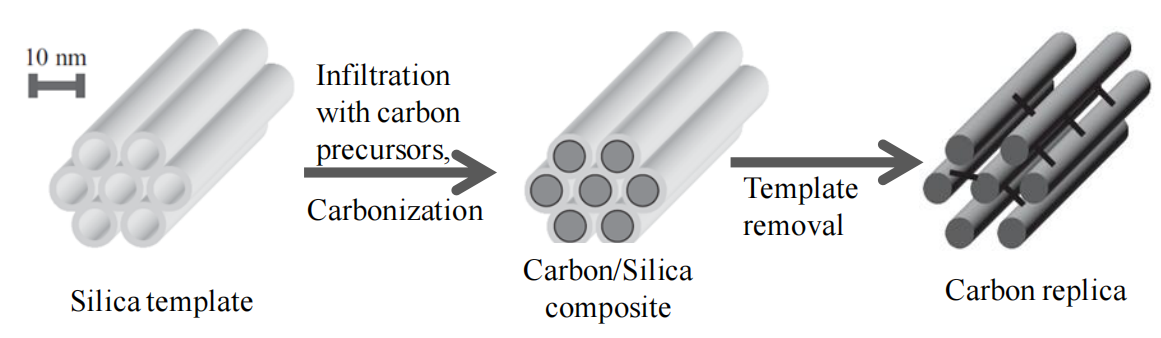
圖5.1. 以有序介孔二氧化硅為模板制備有序介孔碳(OMC)的示意圖[71]。

圖5.2. 負載于碳膠帶上的有序介孔碳樣品場發射掃描電子顯微鏡(SEM)圖像。(a, b) OMC-S、(c, d) OMC-S-HT、(e, f) OMC-A及(g, h) OMC-A-HT的SEM圖像,第一列為低倍率形貌,第二列為高倍率局部細節。部分孔徑≤50 nm的介孔結構以紅色虛線標注。
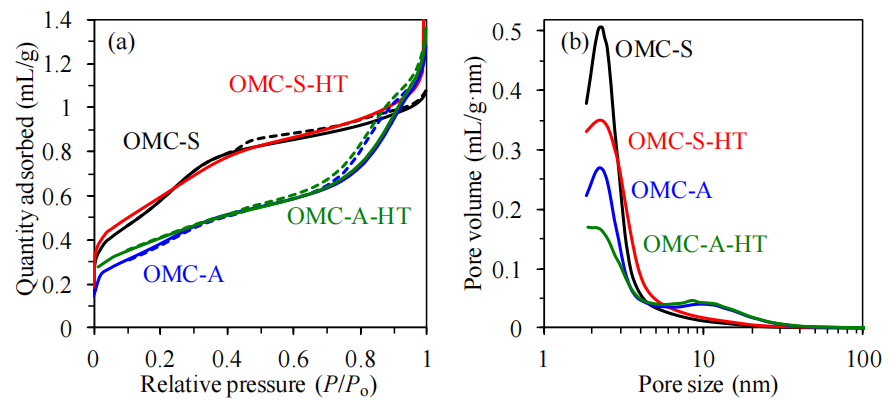
圖5.3. (a) 氮氣吸附(實線)與脫附(虛線)等溫線分析(對比氮氣氣氛中1500°C熱處理2小時前后的有序介孔碳材料OMCs),(b) 基于吸附分支等溫線通過Barrett-Joyner-Halenda (BJH)方法計算的孔徑分布圖(以碳黑標準樣品的t曲線確定氮氣吸附膜的統計厚度)[172]
(注:
測試方法標注:
氮氣吸附-脫附分析是表征多孔材料比表面積、孔徑分布的標準方法,吸附分支反映孔填充過程,脫附分支可指示孔道連通性;
BJH方法基于凱爾文方程計算介孔孔徑分布,需結合標準材料的t曲線校正吸附層厚度以提高精度;
熱處理效應分析:
高溫氮氣處理可能導致OMCs孔壁石墨化或孔道收縮,通過對比熱處理前后的吸附曲線可評估材料結構的熱穩定性;
孔結構關聯性:
孔徑分布集中區域反映模板法合成的介孔特征,而微孔貢獻可能來源于碳骨架內部的缺陷或次級孔隙;
圖表命名規范:
雙縱坐標圖分別表征吸附行為與孔徑分布,符合多孔材料表征的典型呈現形式。)
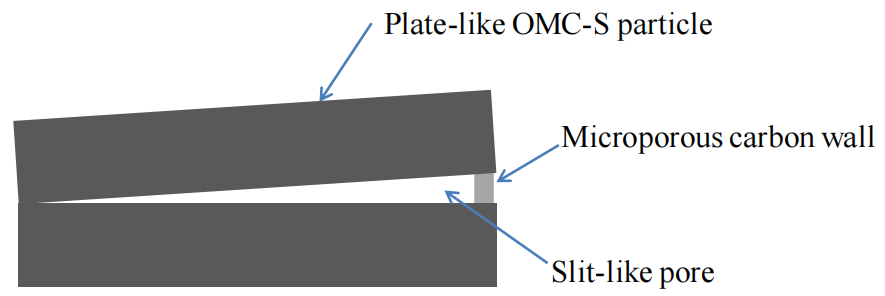
圖5.4. 狹縫型孔道截面結構示意圖(可能由OMC-S顆粒團聚形成,其邊緣及孔口被微孔碳壁封堵)
(注:
結構形成機制:
狹縫型孔道(slit-like pore)的形成可能與OMC-S顆粒的團聚堆積有關,微孔碳壁(microporous walls)兼具結構支撐與傳質限制的雙重作用;
示意圖功能:
通過二維截面簡化三維孔道形貌,輔助理解孔隙結構對吸附/催化傳質路徑的影響(可能需結合后續性能數據關聯分析);
標注規范:
關鍵結構特征(如微孔碳壁、孔道開口)需通過箭頭或文字標注清晰指示,避免歧義。)
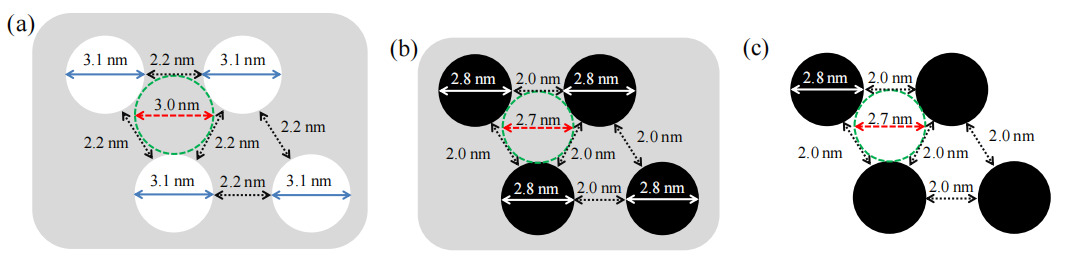
圖5.5. (a) 六方介孔二氧化硅(HMS, 灰色)模板的孔隙(白色圓)與孔壁截面尺寸示意圖(該模板用于合成有序介孔碳OMCs)[49, 132];(b) 碳前驅體填充的HMS模板(a)經高溫(900°C)熱處理后,孔隙內填充碳納米鏈(黑色圓)的截面尺寸示意圖;(c) 移除二氧化硅模板后(b),OMCs孔隙的截面尺寸示意圖。綠色虛線圓(a-c)與其外圍三個圓相切,分別表征HMS模板的二氧化硅壁厚度(a-b)或相鄰碳納米鏈之間的孔隙尺寸(c);箭頭(a-c)表示圓的直徑或兩圓邊緣之間的間隙距離。
注:結合合成機理與文獻規范,
模板法合成邏輯:
HMS的六方有序介孔結構通過浸漬碳前驅體并高溫碳化,最終刻蝕模板獲得反向復刻的OMCs孔道;
結構參數標注:
綠色虛線圓的幾何關系量化孔壁厚度(模板階段)或碳鏈堆積密度(產物階段),箭頭標注則關聯孔道直徑與傳質路徑特征;
熱處理效應:
高溫碳化促使碳前驅體交聯形成連續納米鏈,同時HMS模板的結構穩定性確保孔道形貌精確復制;
示意圖對比意義:
三組示意圖分階段呈現“模板填充-碳化-刻蝕”的合成路徑,直觀揭示OMCs孔道尺寸與模板參數的繼承關系。
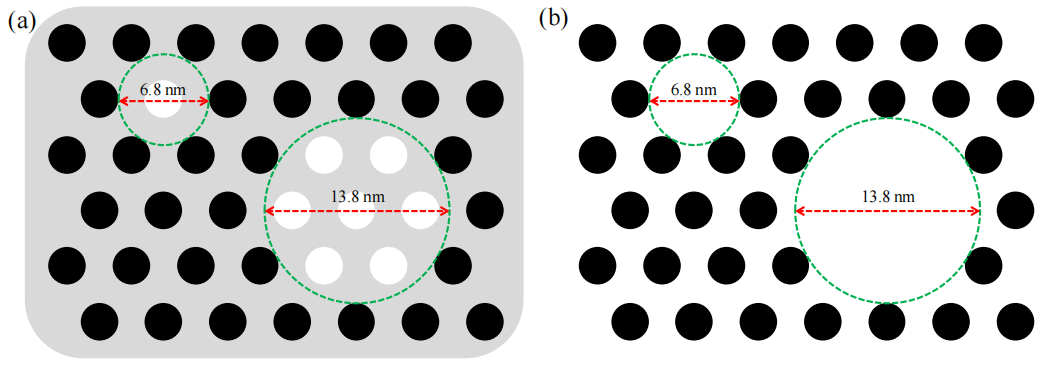
圖5.6. (a) 碳/HMS復合材料經900°C加熱但尚未去除二氧化硅時的截面尺寸示意圖(黑色圓:碳納米鏈;白色圓:未填充孔隙,其尺寸與圖4b一致),(b) 移除二氧化硅后由碳納米鏈包圍的圓柱形孔道示意圖(虛線圓表征碳鏈外圍形成的孔隙)。
注:
結構演化邏輯:
高溫碳化后,HMS模板孔隙內的碳前驅體形成連續碳納米鏈(黑色圓),未填充區域(白色圓)反映模板填充不完全或碳收縮效應;
示意圖關聯性:
(a) 與圖5.5(b)結構參數一致,表明碳/HMS復合界面在高溫下的穩定性;(b)虛線圓對應OMCs的最終孔道形貌,體現模板復刻精度;
虛線圓功能:
虛線圓標記碳納米鏈外圍的潛在孔道邊界,量化碳鏈間距及OMCs孔道尺寸分布(需結合孔徑分析驗證);合成路徑意義:
對比(a)(b)可直觀呈現“碳填充-模板刻蝕”的孔道反轉機制,為調控OMCs孔結構提供可視化依據。
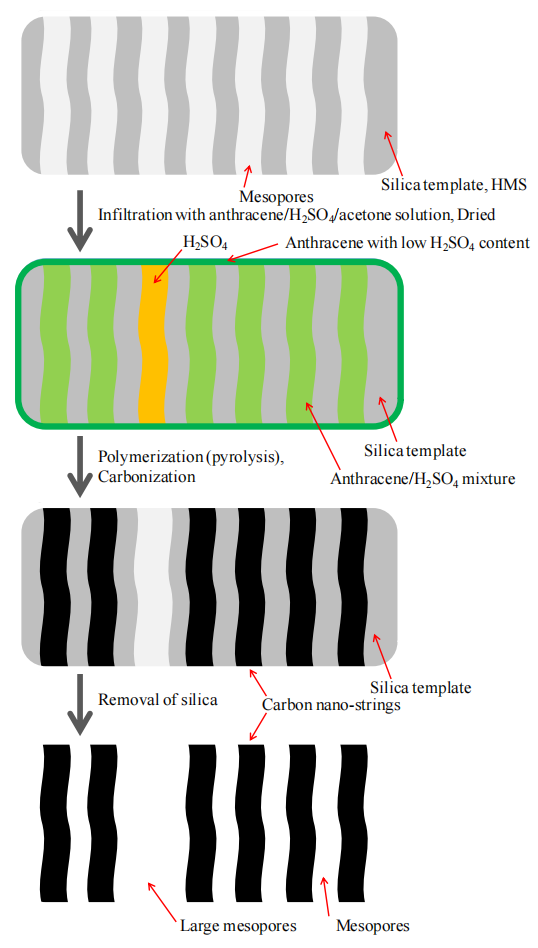
圖5.7. 蒽基有序介孔碳(OMC-A)微觀結構演化機制示意圖。
注:
結構演化機制:
示意圖假設OMC-A的介孔結構形成可能涉及蒽基前驅體的自組裝、模板導向碳化及孔道調控過程,具體路徑可能包含液相浸漬、高溫交聯與模板刻蝕等關鍵步驟;
示意圖關聯性:
需與圖5.5(b)(碳填充模板)及圖5.6(孔道反轉)對比,體現蒽基碳化路徑的獨特性(如分子剛性對孔道有序性的影響);
機制假設性:
“可能機制”需結合原位表征(如SAXS、原位TEM)驗證碳化過程中模板-前驅體界面演變動力學;
合成路徑意義:
通過可視化微觀結構演化,為優化蒽基OMCs的介孔連通性及孔徑均一性提供理論依據。
.png)
圖5.8. 壓片成型的OMC顆粒表面水滴(直徑≈2.7 mm)接觸角動力學(CAK)行為對比圖(氮氣氛圍中1500°C熱處理前后)。標注的“完全干燥時間”指顆粒吸收表面水滴至完全干燥所需時長。
注:結合潤濕性與結構表征數據,
熱處理效應:
高溫處理可能通過石墨化增強碳骨架疏水性,導致接觸角增大及水滴吸收時間延長;
CAK行為關聯性:
熱處理前后接觸角動態變化(如接觸角滯后)可關聯OMCs表面官能團演化與介孔連通性差異;
吸收動力學意義:
“完全干燥時間”反映毛細作用力與孔道潤濕性的競爭關系,量化OMCs表面能及孔隙輸運效率;
可視化對比:
熱處理前后圖像直觀呈現表面親/疏水性調控效果,為功能化OMCs的表面工程提供實驗依據。
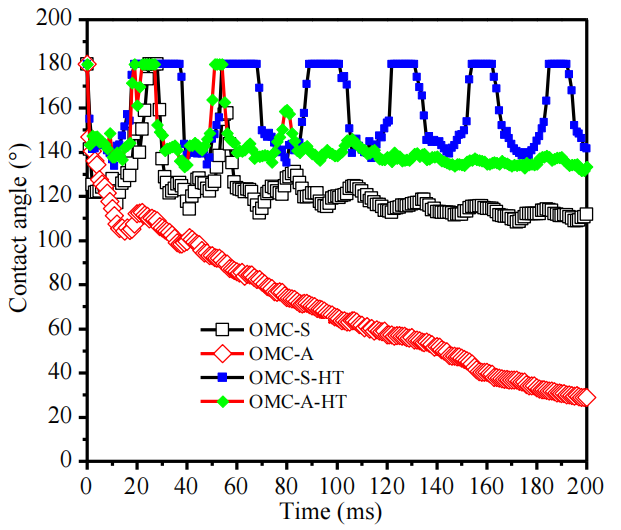
圖5.9. OMC顆粒表面水滴接觸角動力學(CAK)行為對比圖(氮氣氛圍中1500°C熱處理前后,數據源自圖5.8)。
注:
數據量化對比:
結合圖5.8時序圖像,CAK曲線定量表征熱處理前后OMCs表面潤濕性差異(如接觸角衰減速率、完全干燥時間變化);
熱處理效應:
高溫石墨化可能抑制表面極性官能團,降低毛細吸力,導致接觸角滯后增強及水滴吸收動力學延緩;
與前序圖關聯性:
CAK數據需與圖5.6(孔道反轉機制)、圖5.8(潤濕動態可視化)結合,解析表面化學與孔結構對輸運行為的協同影響;
動力學曲線意義:
接觸角-時間曲線斜率反映毛細滲透速率,間接表征OMCs孔道連通性及表面能梯度分布特性。
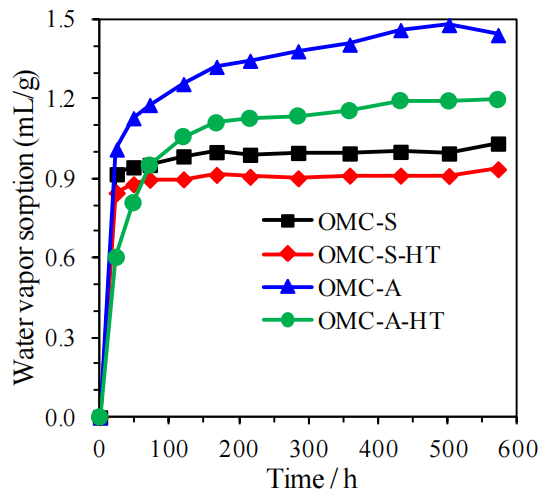
圖5.10. 有序介孔碳材料(OMCs)水蒸氣吸附(VWVS)數據對比圖(氮氣氛圍中1500°C熱處理前后,所有測試均在室溫下進行)。
注:
熱處理對吸附性能的影響:
高溫處理可能通過石墨化減少表面極性官能團并優化孔結構,導致水蒸氣吸附量顯著降低,間接反映材料疏水性增強;
VWVS數據意義:
吸附等溫線特征(如滯后環形態)可關聯OMCs的介孔連通性及表面化學特性,量化熱處理對材料吸/脫附動力學的調控作用;
與潤濕性數據關聯:
需結合圖5.9(接觸角動力學)解析熱處理后表面疏水性與孔道限域效應對水分子輸運行為的協同影響;應用價值:
通過對比熱處理前后吸附性能差異,為設計濕度響應型OMCs或調控其環境穩定性提供關鍵實驗依據。
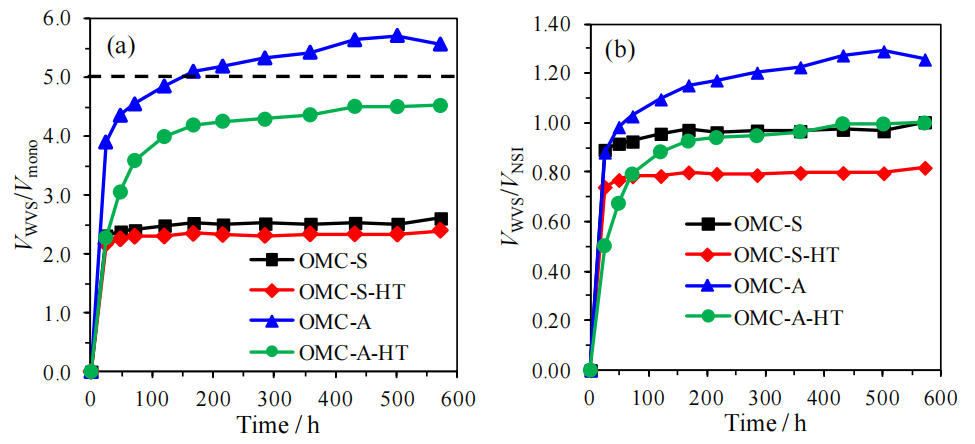
圖5.11. 有序介孔碳材料(OMCs)水蒸氣吸附(VWVS)數據對比圖(氮氣氛圍中1500°C熱處理前后):(a)以比表面積、(b)以孔體積為基準歸一化(數據參考表5.2,來源氮氣吸附等溫線圖5.3a)。

圖5.12.(a) 與水蒸氣在吸附各階段(圖5.10)填充相同孔體積所需的氮氣相對壓力(P/P?,源自圖5.3a氮氣吸附等溫線);(b) 基于凱爾文方程(公式5.2)及炭黑表面氮氣吸附膜的統計厚度(公式5.3),計算的隨時間推移被水完全填充的孔尺寸估算值(對應圖5.10中各吸附階段數據點)。
注解:
(a)氮氣與水蒸氣填充孔體積的對比:
通過對比氮氣(非極性)與水蒸氣(極性)填充相同孔體積所需的相對壓力差異,可間接反映OMCs孔道表面化學性質(極性/非極性)對吸附機制的調控作用;
(b)孔徑估算方法:
· 凱爾文方程:
rK=−2γVmRTln?(P/P0)
rK?=
RTln(
P/
P0?)−2
γVm??
用于關聯孔填充壓力與孔徑(r<sub>K</sub>為凱爾文半徑),結合氮氣吸附膜的統計厚度修正,可更準確推算實際孔尺寸分布;
· 數據意義:
動態孔徑估算結果可揭示熱處理后孔道連通性優化或表面疏水性增強對水分子漸進填充行為的抑制效應。
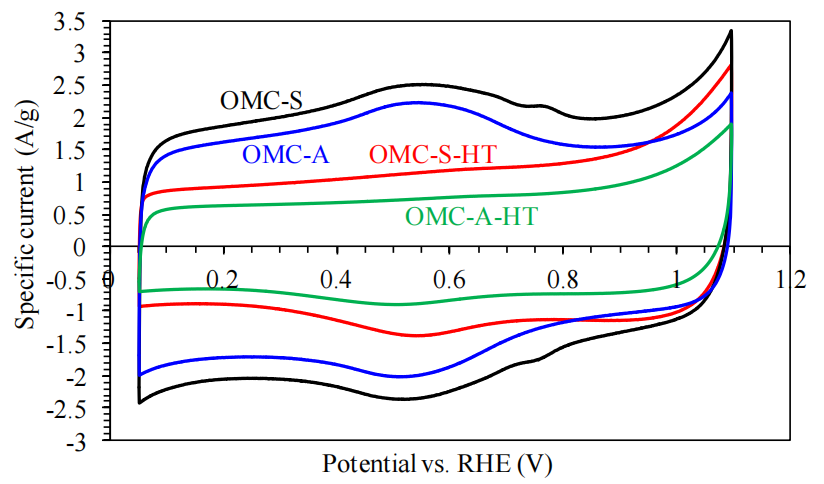
圖5.13. 熱處理前后有序介孔碳材料(OMCs)的循環伏安曲線(CVs)(測試條件:氮氣飽和的0.5 M H?SO?溶液,掃描速率10 mV/s)。
注解:
循環伏安法(CV)測試意義:
通過對比熱處理前后OMCs的CV曲線,可表征材料電化學活性比表面積(ECSA)、表面官能團氧化還原特性及電荷存儲能力的變化;
典型碳材料在酸性電解液中,CV曲線的矩形度反映雙電層電容行為,氧化還原峰則與表面含氧官能團(如羧基、羥基)相關;
熱處理效應分析:
電化學活性變化:熱處理后CV曲線矩形度增強,可能因石墨化程度提高導致雙電層電容主導,同時表面含氧官能團減少(氧化還原峰減弱);
電荷傳輸優化:若熱處理后電流響應增大,可能與孔道連通性改善、導電性增強及雜質去除相關;
參數說明:
掃描速率:10 mV/s的低掃速可減少極化效應,更接近準平衡態下的電化學行為表征;
電解液選擇:0.5 M H?SO?為常用酸性電解液,適用于碳基材料的雙電層電容評估。
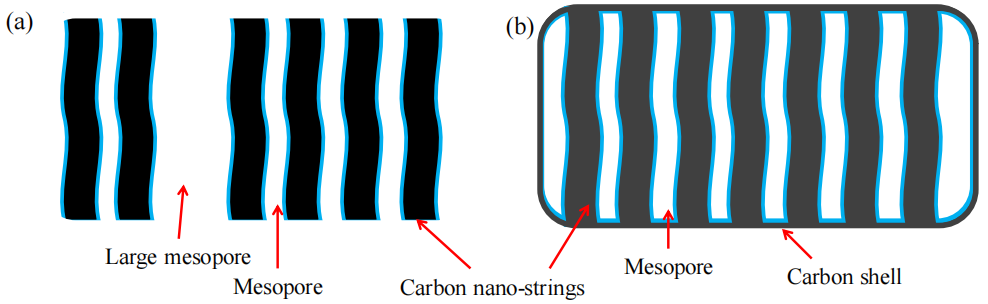
圖5.14. OMC-A與OMC-S截面微觀結構示意圖
(a) OMC-A:彎曲的黑色條帶表示OMC納米弦;(b) OMC-S:黑色矩形框表示包裹OMC-S納米弦的碳層(或碳殼)。藍色層代表極性或親水性碳表面。相較于OMC-A納米弦(表5.2),OMC-S的碳殼及納米弦含有更多微孔。OMC-A(a)中的大中孔對應于圖5.6b中示意性標注的6.8 nm孔徑結構。
注解:
結構差異表征:
OMC-S碳殼作用:碳殼包裹納米弦可增強結構穩定性,同時其表面極性區域(藍色)可能促進水分子吸附與離子傳輸;
微孔分布效應:OMC-S中更高的微孔密度可能提升電化學活性位點密度,但或導致傳質阻力增加;
大中孔功能:
OMC-A中6.8 nm介孔的形成與模板劑(如二氧化硅硬模板)的尺寸調控直接相關,其開放孔道有利于電解液的快速滲透。
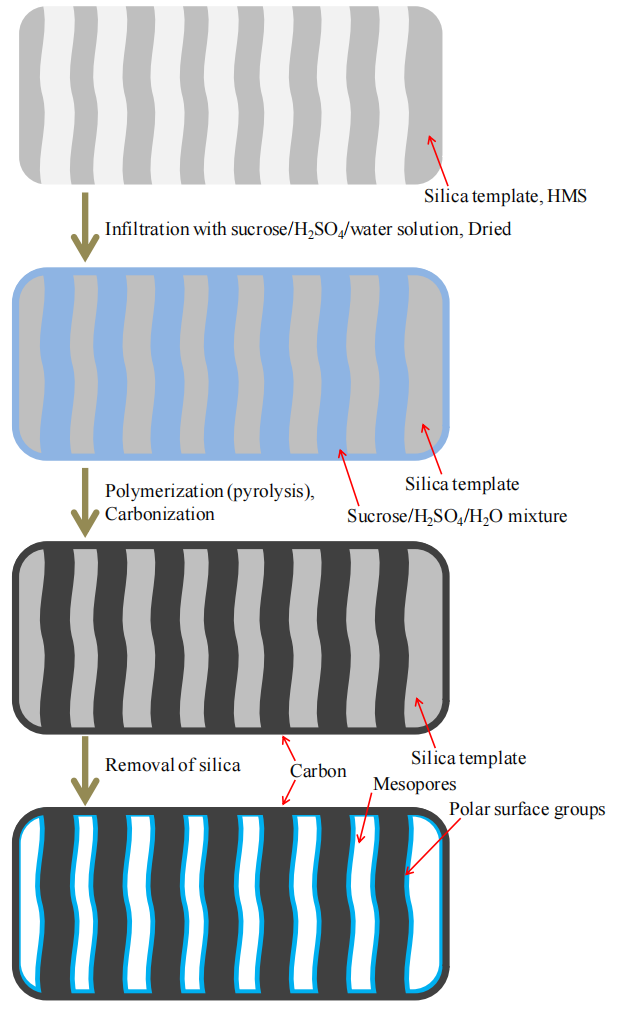
圖5.15. 蔗糖基有序介孔碳材料(OMC-S)微觀結構演化機制的示意圖
注解:
模板導向機制:
OMC-S的介孔結構形成可能基于模板劑(如介孔二氧化硅)的納米限域效應:
步驟1:蔗糖前驅體溶液滲透至模板介孔中;
步驟2:高溫碳化過程中,蔗糖轉化為碳骨架并復制模板的孔道結構;
步驟3:模板去除后,殘留的碳骨架形成高度有序的介孔網絡;
表面化學調控:
蔗糖碳化過程中可能生成極性含氧官能團(如羥基、羧基),賦予OMC-S表面親水性(對應圖5.14b中藍色親水層);
微孔-介孔協同形成:
介孔主框架:由模板劑尺寸直接調控(如6.8 nm介孔對應圖5.6b);
微孔結構:源自碳化過程中前驅體分子交聯及揮發分的釋放,其密度受碳化溫度與時間影響(表5.2);
結構穩定性機制:
碳殼包裹納米弦結構(圖5.14b)可能通過抑制碳骨架收縮或塌陷,維持高溫處理后的孔結構完整性。
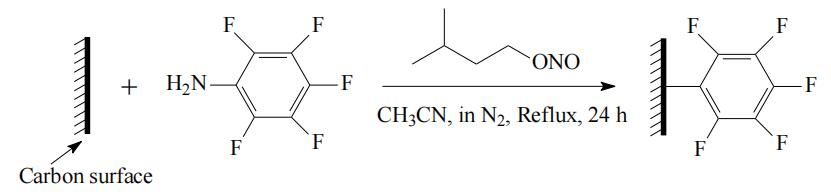
示意圖6.1. 碳表面五氟苯基(-PhF5)基團功能化示意圖
注解(基于常規學術背景推測,僅供參考):
功能化機理:
五氟苯基基團通過化學修飾(如共價鍵接枝或物理吸附)引入碳表面,調控其表面化學性質與反應活性;可能的反應路徑包括自由基偶聯、芳基化反應或電化學沉積等;
功能化效應:
疏水性增強:-PhF5基團的強疏水特性可降低碳表面親水性,適用于非極性環境中的催化或吸附應用;
電子結構調控:氟原子的強電負性可能改變碳材料表面電子分布,影響其導電性或催化選擇性;
表征方法:
功能化后的碳表面可通過X射線光電子能譜(XPS)、接觸角測試或拉曼光譜驗證基團接枝效果。
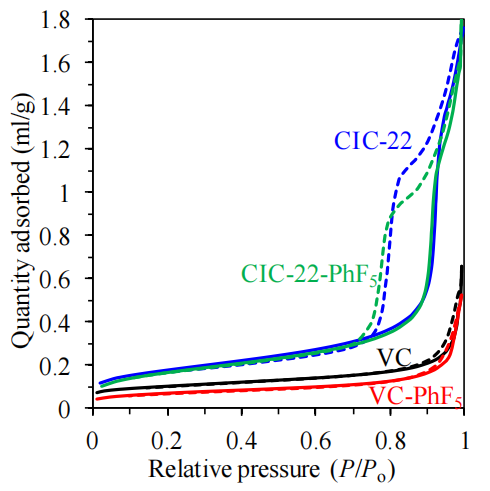
圖6.1. 基于重氮鹽還原反應(示意圖6.1)實現五氟苯基(-PhF5)基團表面功能化的VC與CIC-22材料功能化前后的N2吸附(實線)-脫附(虛線)等溫線
注解:
等溫線特征解析:
功能化前后等溫線滯后環形狀變化可反映材料孔徑分布與表面化學性質的改變(如微孔填充效應或介孔毛細凝聚行為);
功能化對比:
-PhF5基團引入可能導致比表面積下降(因表面基團占據孔隙)或特定孔道選擇性修飾(如疏水基團抑制氮氣分子在微孔內的吸附);
分析方法:
通過BET模型計算比表面積,結合DFT/NLDFT方法解析孔徑分布差異,量化功能化對孔結構的影響。
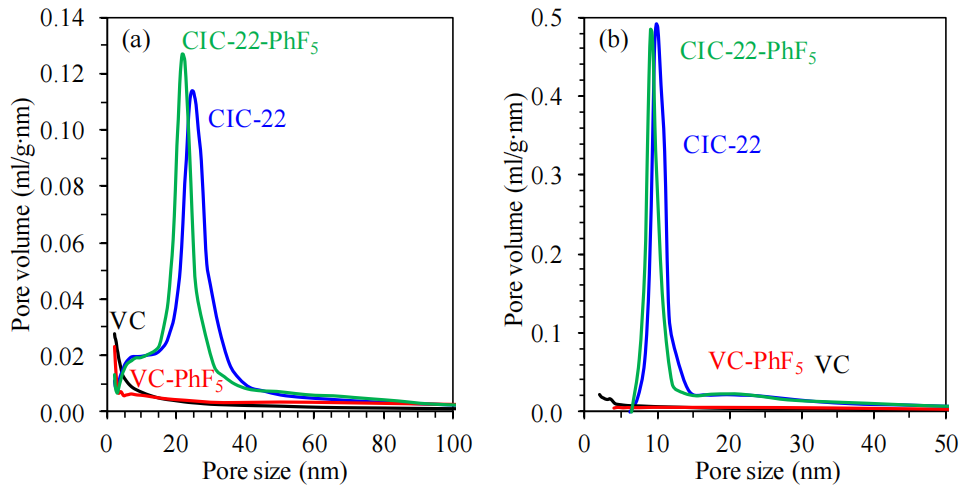
圖6.2. 碳材料表面功能化前后的孔徑分布(基于N2吸附-脫附等溫線數據計算)
(a)吸附支、(b)脫附支數據通過Barrett-Joyner-Halenda (BJH) 方法計算,并以炭黑t-曲線為標準確定氮氣吸附膜的統計厚度。
注解(結合常規分析補充):
功能化對孔徑的影響:
微孔減少:-PhF5基團接枝可能覆蓋部分微孔表面(如功能化后<2 nm孔隙占比下降),導致有效孔徑分布向介孔偏移;
介孔結構變化:脫附支計算的孔徑分布(圖6.2b)更敏感于孔喉尺寸,功能化后滯后環位移(圖6.1)可能反映孔道連通性或孔口阻塞效應;
BJH方法適用性:
BJH基于圓柱孔假設,適用于介孔分析(2-50 nm),但對微孔(<2 nm)精度有限,需結合DFT模型補充(如正文表6.1);
t-曲線選擇的意義:
炭黑t-曲線適用于非石墨化碳材料,其統計厚度模型可減少因表面化學差異(如功能化引入氟原子)導致的吸附層厚度計算誤差。
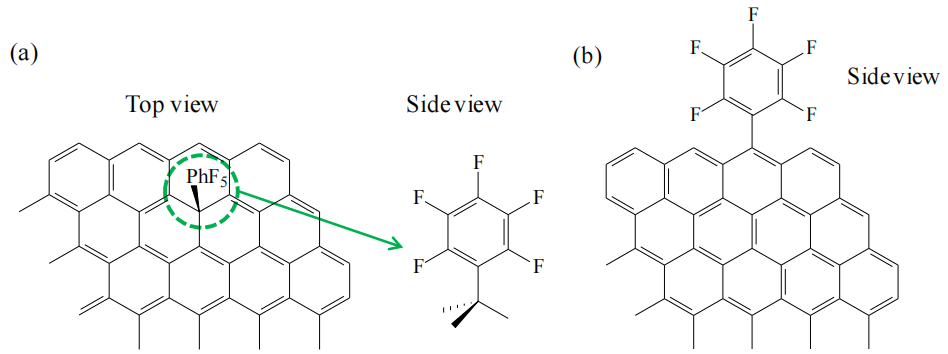
示意圖6.2. -PhF5基團與碳表面間C-C單鍵的推測連接方式示意圖
(a) 平面石墨烯表面;(b) 石墨烯邊緣表面(頂面)。
注解:
功能化位點差異:
平面石墨烯(圖a):C-C單鍵可能通過芳環平面上的sp²碳與-PhF5基團形成定向共價連接,受表面π電子云分布影響;
石墨烯邊緣(圖b):邊緣sp³雜化碳或含氧官能團(如羧基)更易與-PhF5發生化學鍵合,反應活性通常高于平面區域;
鍵合方式影響:
平面鍵合可能保留石墨烯的本征導電性,而邊緣鍵合可能引入局部結構畸變,影響電荷傳輸與機械穩定性;
表征驗證:
可通過掃描隧道顯微鏡(STM)觀測平面鍵合位點的原子排布,或通過同步輻射X射線吸收譜(XAS)區分sp²/sp³雜化碳的鍵合狀態。
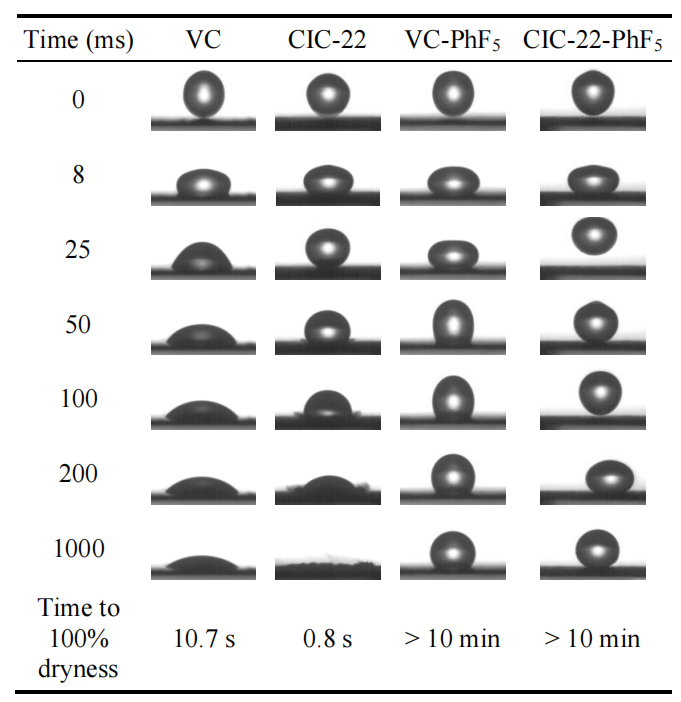
圖6.3. 五氟苯基(-PhF5)基團表面功能化前后碳素顆粒表面水滴沉積的時序圖像
(最后一行)完全干燥時間指碳丸完全吸收其表面水滴所需的時間。
注解:
表面潤濕性變化:
功能化前:原始碳顆粒可能因表面含氧官能團(如羥基、羧基)呈現親水性,促進水滴快速吸收;
功能化后:-PhF5基團的強疏水性(氟原子電負性效應)可能顯著降低表面能,延長水滴吸收時間;
吸收動力學差異:
干燥時間的變化可能與表面孔徑結構及化學異質性相關:疏水化修飾可能抑制微孔內的毛細作用,但介/大孔區域的物理吸附仍可能主導吸收速率;
表征關聯性:
可通過接觸角測量量化潤濕性轉變,并與XPS表面元素分析(F含量)關聯,驗證功能化程度對吸收行為的影響。
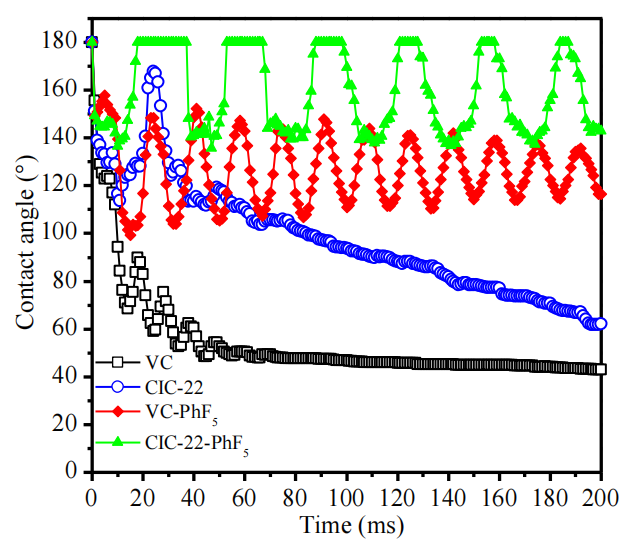
圖6.4. 碳丸表面經–PhF5基團功能化前后水滴沉積的接觸角動態變化(CAK)
接觸角為180°表示水滴已從樣品表面完全反彈(例如圖6.3中CIC-22-PhF5樣品在t=25 ms時的狀態)。
注解(結合潤濕性動力學分析):
接觸角極值意義:
180°超疏水特征:表明表面具有類“荷葉效應”(Cassie-Baxter態),液滴因表面粗糙微結構與低表面能(-PhF5修飾)而無法浸潤,僅通過氣墊層接觸,最終彈離表面;
動態響應差異:原始碳表面接觸角隨時間衰減(如親水表面液滴鋪展),而功能化后接觸角維持高值或快速達180°,反映疏水穩定性增強;
CAK曲線解讀:
反彈時間(如25 ms)可關聯表面粘附力:超疏水狀態下,液滴滯留時間越短,表明固-液界面粘附能越低,表面自清潔潛力越高;
與形貌表征的關聯性:
需結合SEM/AFM觀測表面粗糙度,驗證–PhF5修飾是否誘導微納分級結構(如納米突起或孔道),協同化學疏水性實現超疏水行為。
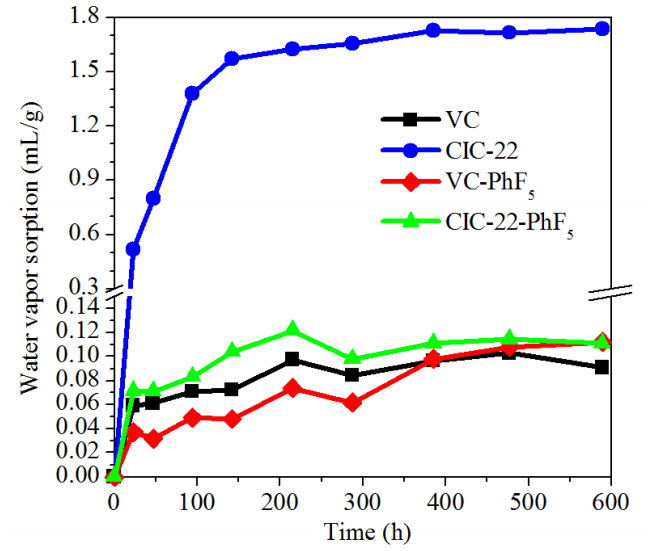
圖6.5. 五氟苯基(-PhF5)基團表面功能化前后VC與CIC-22材料在室溫水蒸氣環境中的水蒸氣吸附(WVS)數據
注解(結合吸附行為分析):
疏水改性效應:
-PhF5基團的引入顯著降低材料表面能,氟原子高電負性與低極性抑制水分子吸附,導致功能化后樣品吸附量下降;這一現象與含氟化合物在表面改性中普遍表現出的疏水特性一致;
吸附動力學差異:
原始碳材料(如CIC-22)因表面含氧官能團(羥基、羧基等)的親水性,可能通過氫鍵作用加速水蒸氣吸附;而功能化后,化學惰性-PhF5基團阻礙了水分子與表面的直接接觸,吸附速率降低;
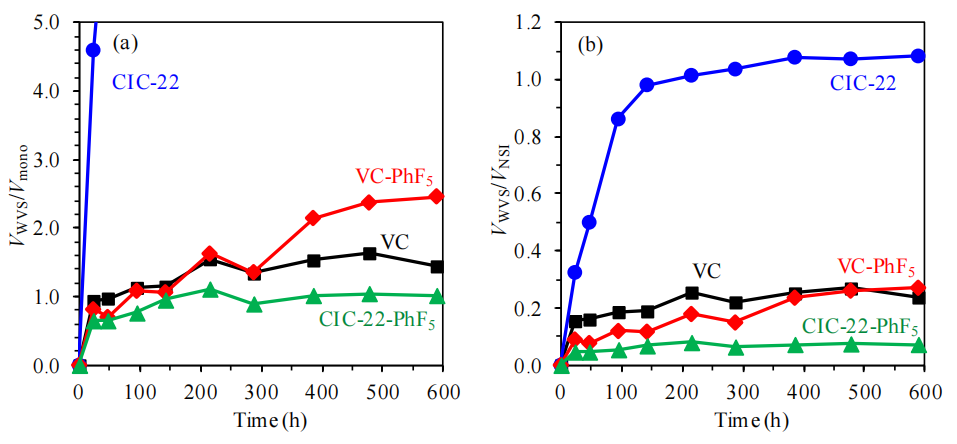
圖6.6. VC與CIC-22材料表面功能化前后的水蒸氣吸附數據(VWVS,源自圖6.5),分別基于(a)比表面積和(b)孔體積(對應表6.1中的SBET與SNSI數據)進行歸一化處理,數據來源于氮氣吸附等溫線(圖6.1)。
注解(結合吸附性能量化分析):
歸一化意義:
通過比表面積(SBET)與孔體積(SNSI)歸一化,可區分表面化學修飾(-PhF5)與物理結構(孔隙率)對水蒸氣吸附的貢獻,避免因比表面積差異導致的數據偏差;
化學修飾主導性:
若歸一化后吸附量仍顯著下降,表明-PhF5的疏水效應超越物理結構影響,表面化學性質成為吸附抑制的主因;
結構-功能協同效應:
需結合表6.1的孔隙參數,分析微孔堵塞或介孔覆蓋率是否導致歸一化吸附量變化,驗證化學修飾對活性位點的屏蔽程度;
數據對比方法學:
氮氣吸附(非極性分子)與水蒸氣吸附(極性分子)的差異可凸顯表面極性修飾的影響,為疏水材料設計提供量化依據。
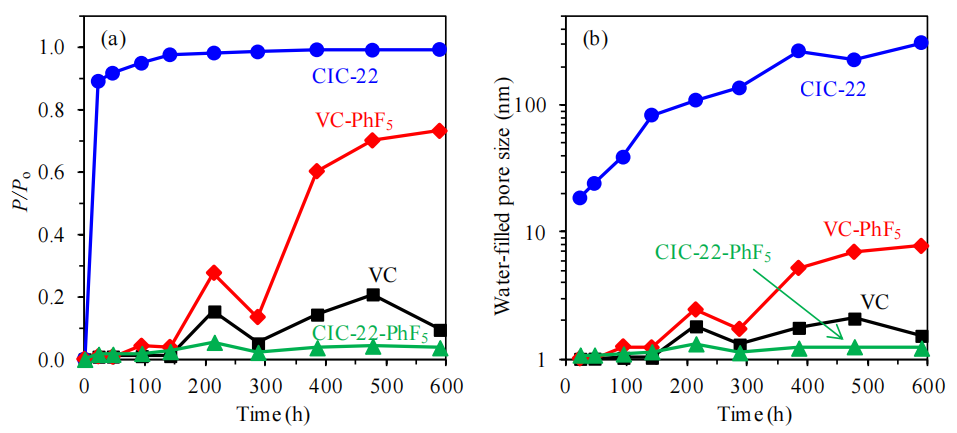
圖6.7. (a) 填充與水蒸氣吸附(圖6.5)各階段相同孔體積所需的氮氣(圖6.1)冷凝相對壓力(P/Po);(b) 根據(a)中對應的相對壓力值,通過開爾文方程(公式5.2)及炭黑表面吸附氮氣膜的統計厚度(公式5.3)[172]計算得出的隨時間推移被水填充的孔隙尺寸估計值(對應圖6.5中各時間點),具體方法詳見第5章描述。
注解:
相對壓力關聯性:
氮氣(非極性)與水蒸氣(極性)的吸附壓力差異反映了孔隙表面化學性質的調控作用:疏水改性(-PhF5)可能通過降低表面極性,抑制氮氣冷凝所需壓力,間接驗證功能化對孔隙潤濕性的影響;
開爾文方程適用性:
水蒸氣吸附中,因氫鍵與表面極性作用顯著,經典開爾文方程(基于非極性分子假設)可能高估實際孔徑,需引入極性修正項或對比氮氣吸附模型以校準計算誤差;
動態填充序列:
隨時間變化的孔徑分布曲線可揭示水分子優先填充微孔(強吸附位)或介孔(毛細凝聚)的路徑,結合吸附等溫線滯后環分析,可區分表面吸附與體相冷凝的貢獻;
功能化干擾機制:
若功能化后相同壓力下填充孔徑顯著減小,表明-PhF5基團可能通過物理堵塞(微孔入口)或化學屏蔽(覆蓋活性位點)改變孔隙可及性,需結合XPS或孔徑分布數據驗證。
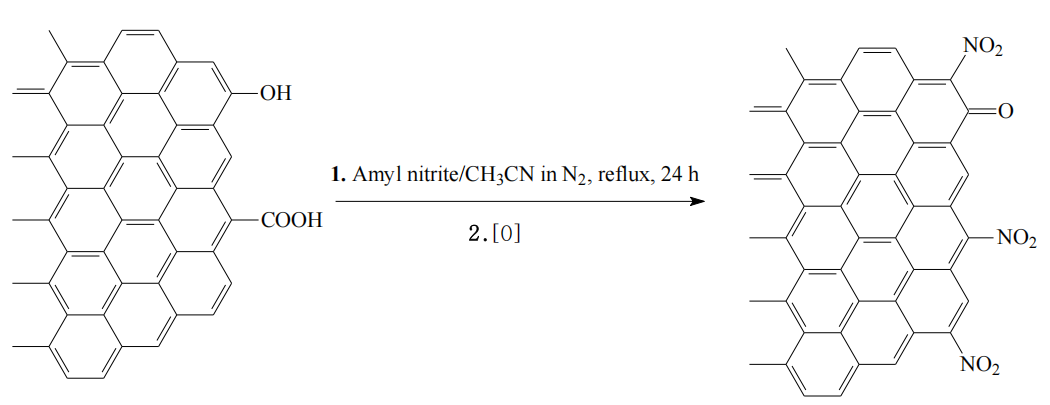
示意圖6.3. 碳表面在功能化反應(示意圖6.1)中與亞硝酸戊酯的可能反應路徑
注解:
反應類型推測:
亞硝酸戊酯作為硝化試劑,可能與碳表面活性位點(如缺陷位點、邊緣碳原子)發生硝化反應(引入硝基-NO?)或酯化反應(生成C-O-NO鍵),具體路徑取決于表面電子狀態及溶劑環境;
反應位點選擇性:
sp²/sp³雜化碳的化學活性差異可能導致反應優先發生在石墨烯缺陷區(如五元環、七元環)或邊緣羧酸基團附近,需結合XPS或FTIR表征驗證修飾基團類型;
反應條件影響:
溶劑極性、溫度及反應時間可能調控亞硝酸戊酯的分解速率(釋放NO?或自由基),進而影響功能化產物的分布(如單取代或多取代修飾);
副反應可能性:
若反應體系中存在微量水或酸性條件,亞硝酸戊酯可能水解生成硝酸,引發碳骨架氧化(生成羧酸基團),需通過TGA或元素分析檢測表面氧含量變化。
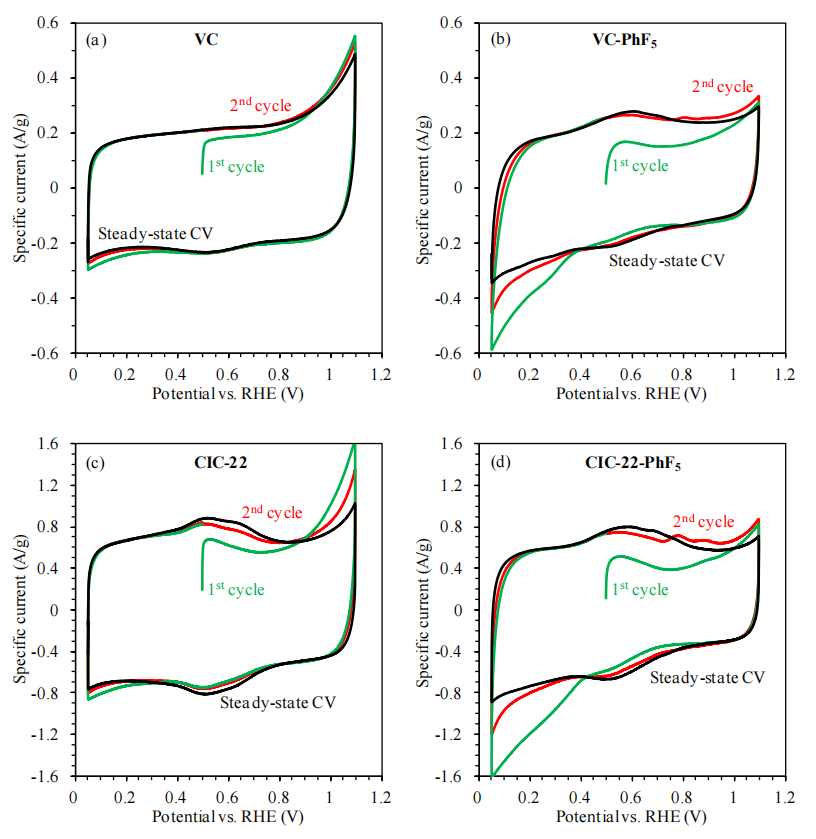
圖6.8. VC、VC-PhF5、CIC-22及CIC-22-PhF5材料在氮氣飽和的0.5 M H?SO?溶液中(掃描速率10 mV/s)的循環伏安曲線(CVs)
注解(結合電化學性能分析):
反應可逆性表征:
CV曲線的對稱性及氧化還原峰電位差(ΔE)可反映電極反應的可逆性:ΔE接近59 mV(單電子轉移)表明可逆性良好,ΔE增大則提示反應動力學受限或表面修飾導致電子轉移阻力增加;
表面修飾影響:
-PhF5功能化可能通過疏水效應抑制質子(H?)在電極表面的擴散,導致VC-PhF5與CIC-22-PhF5的氧化還原峰電流降低,需結合電化學阻抗譜驗證界面電荷轉移阻力變化;
活性位點可及性:
對比功能化前后CV曲線積分電荷量差異,可評估表面修飾對活性位點(如含氧官能團或缺陷位點)的屏蔽作用,若電荷量顯著下降則表明功能化基團堵塞了電化學反應活性區域;
電解質滲透路徑:
氮氣飽和條件下的酸性環境可能影響孔隙內電解質浸潤性,若功能化后CV曲線雙電層電容顯著降低,提示疏水基團阻礙了孔隙內部電解質的有效滲透。
創新點總結
新型碳材料設計與合成
開發了膠體印跡碳(CIC)與有序介孔碳(OMC)兩種結構可控的碳載體材料,其中CIC因其大孔徑和堅固孔壁結構,突破了傳統碳載體(如OMC)因孔徑過小導致的氣體傳輸限制,為催化劑層的水-氣協同管理提供了新選擇。
潤濕性調控方法的創新
提出通過熱處理(提升結晶度與導電性)與表面氟化(化學修飾)的雙重策略調控碳材料疏水性,顯著優化了材料的水吸附特性。這一復合改性方法在疏水性調控效率上優于單一手段,且兼顧了材料導電性與結構穩定性。
多維度潤濕性表征技術
結合接觸角動力學(CAK)與水蒸氣吸附(WVS)實驗,從動態潤濕行為(宏觀)和吸濕能力(微觀)雙重視角定量評估材料潤濕性,彌補了傳統單一接觸角測量的局限性,為燃料電池材料的水管理研究提供了更全面的分析手段。
結構-性能關聯的新發現
揭示了碳材料孔徑大小與孔壁機械強度對水傳輸和氣體擴散的協同影響機制,提出CIC的“大孔徑+高穩定性”特性可同時緩解燃料電池的水淹(陰極)與膜干涸(陽極)問題,為高性能載體材料的設計提供了理論依據。
本文研究了PEMFC中碳材料的潤濕性及其調控方法。通過合成不同孔徑的CIC和OMC,并采用熱處理和表面氟化等方法對其潤濕性進行了調控。結果表明,熱處理和表面氟化均能有效提高碳材料的疏水性,而CIC由于其較大的孔徑和堅固的孔壁結構,在PEMFC應用中顯示出更大的潛力。未來的研究將進一步探索碳材料的表面改性方法及其在PEMFC中的實際應用效果。研究通過材料合成、表面改性及跨尺度表征的集成創新,提出了碳載體潤濕性優化與結構設計協同調控的新路徑,對提升質子交換膜燃料電池的耐久性與輸出性能具有重要指導意義。
https://prism.ucalgary.ca
轉自《石墨烯研究》公眾號










